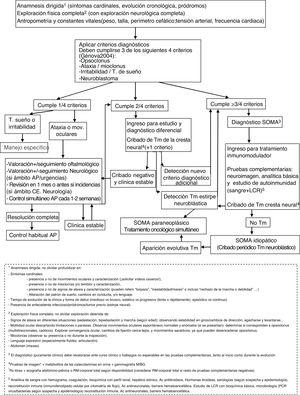
La histiocitosis de células de Langerhans (HCL) es actualmente considerada una neoplasia inflamatoria rara de origen mieloide que afecta a diferentes órganos y sistemas, de presentación clínica heterogénea. La HCL neurodegenerativa (HCL-ND) es una entidad tardía y poco frecuente, caracterizada por un cuadro clínico-radiológico progresivo que supone una importante morbilidad, con una evolución variable e impredecible. Históricamente fue considerada una secuela de probable etiología paraneoplásica, con tratamientos poco efectivos. La afectación multisistémica, la presencia de diabetes insípida central y la afectación ósea orbitaria y/o de la base de cráneo se consideran factores de riesgo asociados al desarrollo de la HCL-ND. El diagnóstico precoz, eminentemente clínico y radiológico, apoyado por estudios electrofisiológicos y neurocognitivos, permitiría un tratamiento temprano, posiblemente clave para revertir o ralentizar la evolución. En los últimos años, el descubrimiento de la presencia de mutaciones activadoras de la vía dependiente de la proteína quinasa de activación mitogénica (MAPK) y el desarrollo de modelos animales en ratón han permitido redefinir esta entidad como una forma de enfermedad activa de HCL que produce neuroinflamación y neurodegeneración, suponiendo un cambio en el conocimiento de la etiopatogenia, identificando nuevos factores de riesgo y posibles tratamientos dirigidos.