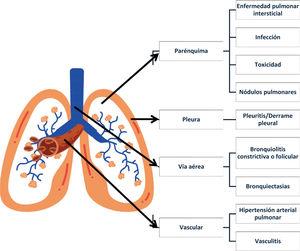
Las enfermedades pulmonares intersticiales (EPI), son un grupo heterogéneo de trastornos, caracterizados por inflamación y/o fibrosis, que afectan el intersticio pulmonar. Históricamente, se basaban en características histopatológicas de biopsias pulmonares, pero con el avance de la tomografía computarizada de alta resolución (TCAR), el enfoque ha cambiado, permitiendo identificar patrones de enfermedad que sugieren distintas etiologías. La clasificación de las EPI ha evolucionado, y actualmente se clasifican según etiología, patrones histopatológicos y/o radiológicos y curso evolutivo. Las neumonías intersticiales idiopáticas (NII) son un grupo de EPI de origen desconocido, y la fibrosis pulmonar idiopática (FPI) es la más común dentro de las NII. Se ha desarrollado el concepto de enfermedad pulmonar intersticial fibrosante progresiva (EPI-FP), que describe un fenotipo de EPI con progresión a pesar del tratamiento. El diagnóstico se basa en una historia clínica detallada, examen físico, pruebas de función pulmonar, serología autoinmune y técnicas de imagen como la TCAR. El tratamiento varía según la etiología y puede incluir terapia inmunomoduladora o antiinflamatoria, terapia modificadora de la enfermedad con agentes antifibróticos y, en casos avanzados, trasplante pulmonar. La atención médica debe ser holística, incluyendo rehabilitación pulmonar, prevención de infecciones y oxigenoterapia.
El objetivo de este artículo es realizar una actualización clínica de las enfermedades pulmonares intersticiales y comentar los avances en las estrategias diagnósticas y terapéuticas.